Accurate annotation of all protein-coding sequences (CDSs) is an essential prerequisite to fully exploit the rapidly growing repertoire of completely sequenced prokaryotic genomes. However, large discrepancies among the number of annotated CDSs, missed functional short ORFs, and overprediction of spurious ORFs represent serious limitations.

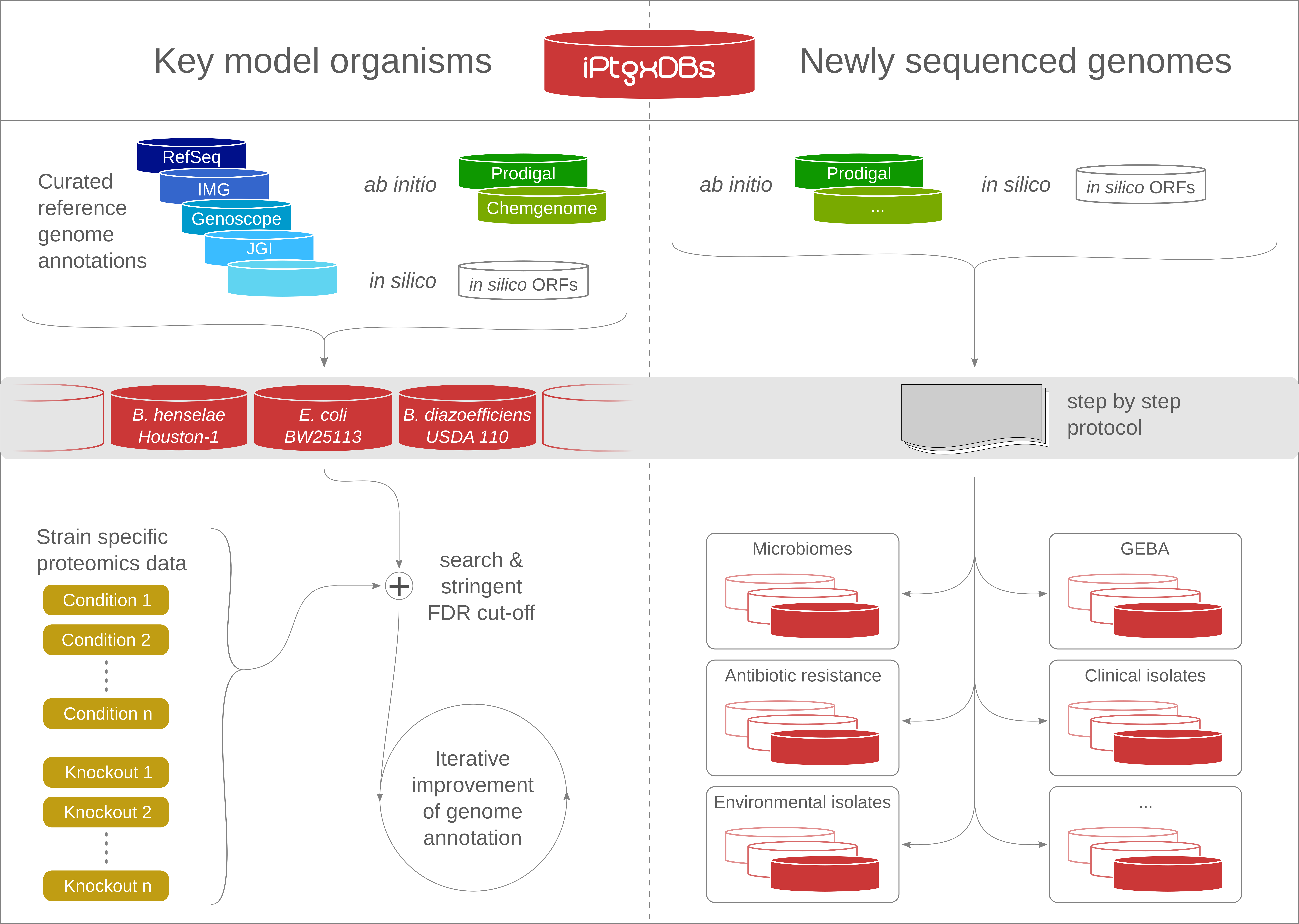
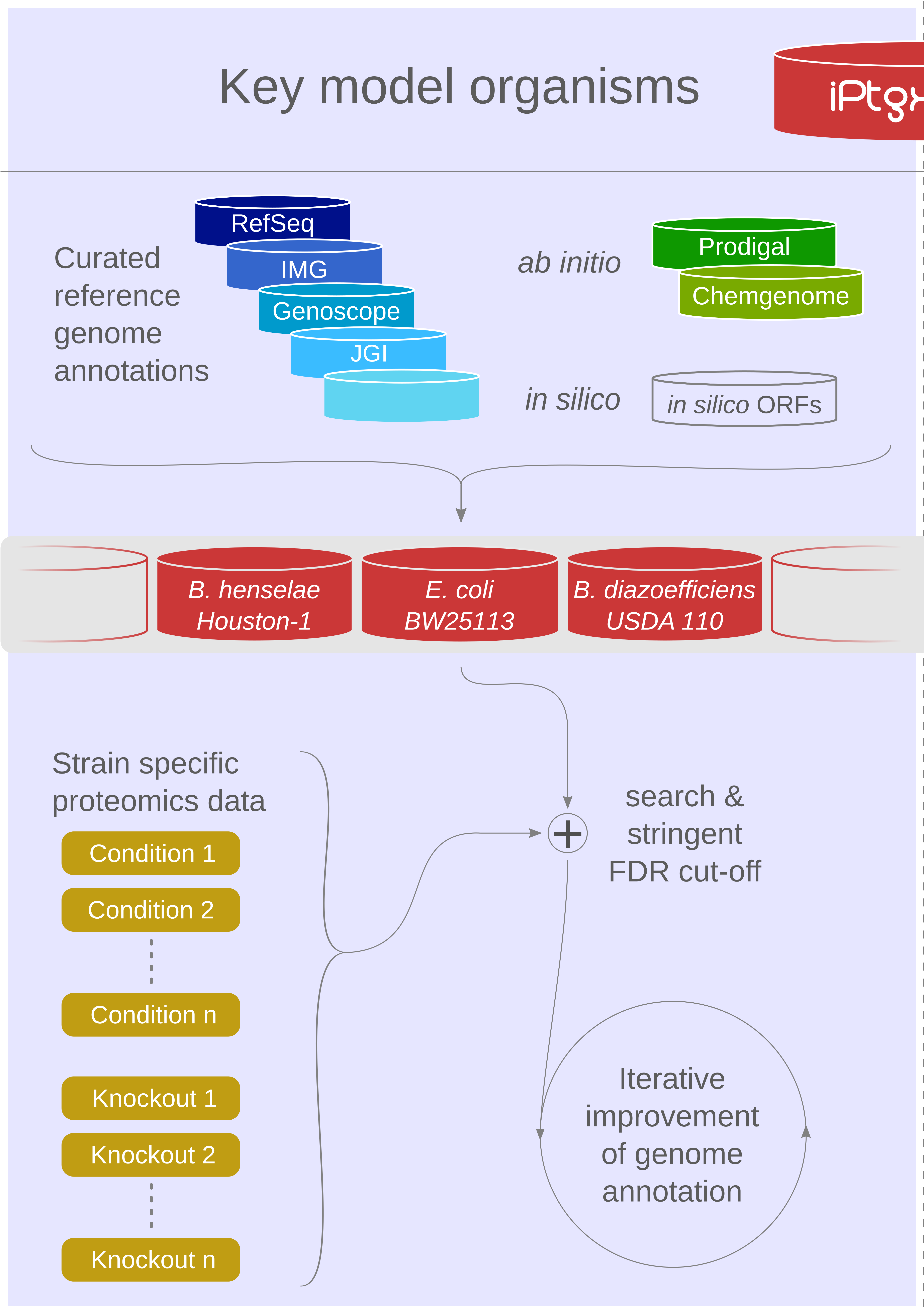
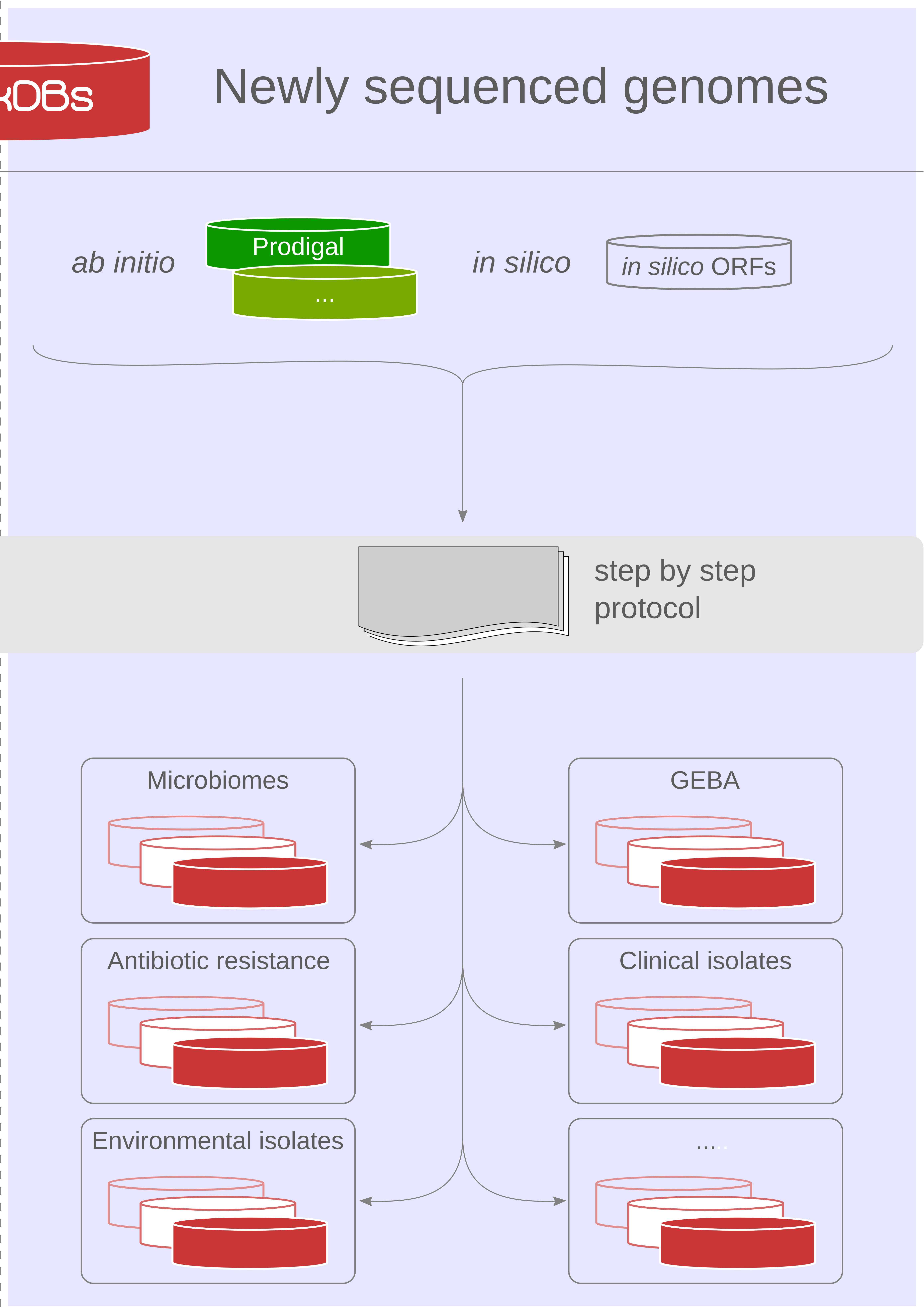
Our proteogenomics [1,2] strategy for accurate and complete genome annotation consolidates CDSs from multiple reference annotation resources, ab initio gene prediction algorithms and in silico ORFs in an integrated proteogenomics database (iPtgxDB) that covers the entire protein-coding potential of a prokaryotic genome [3].

iPtgxDBs address an unmet need of the research community, i.e. an open source DB that provides integrated annotations, predictions and a six-frame translation for one respective genome sequence in an easily usable format, both as a search DB (FASTA format) with informative identifiers and a GFF file that integrates all annotations and identifiers. The search DB is highly informative: by extending the PeptideClassifier concept of unambiguous peptides [4], close to 95% of the mass spectrometry-identifiable peptides imply one distinct protein, largely simplifying downstream analysis and overcoming the need to dis-entangle protein groups implied by shared peptides [5].

Using our precomputed iPtgxDBs or by generating their own, researchers can swiftly identify novel short ORFs (sORFs; [6]), start sites or wrongly annotated pseudogenes.