Below are several examples of novel ORFs, start sites or wrongly annotated pseudogenes, in this case identified in the genome of the alpha-proteobacterium B. henselae strain Houston-1 (38.2% GC). For illustration, a single frame of the forward/reverse strand with possible start (green) and stop codons (red) is shown, along with annotations and experimental evidence (spectral counts scaled from 0 to 20).
In addition, we show two examples (example 1 and example 2) that highlight the critical importance of having the correct genome sequence available.
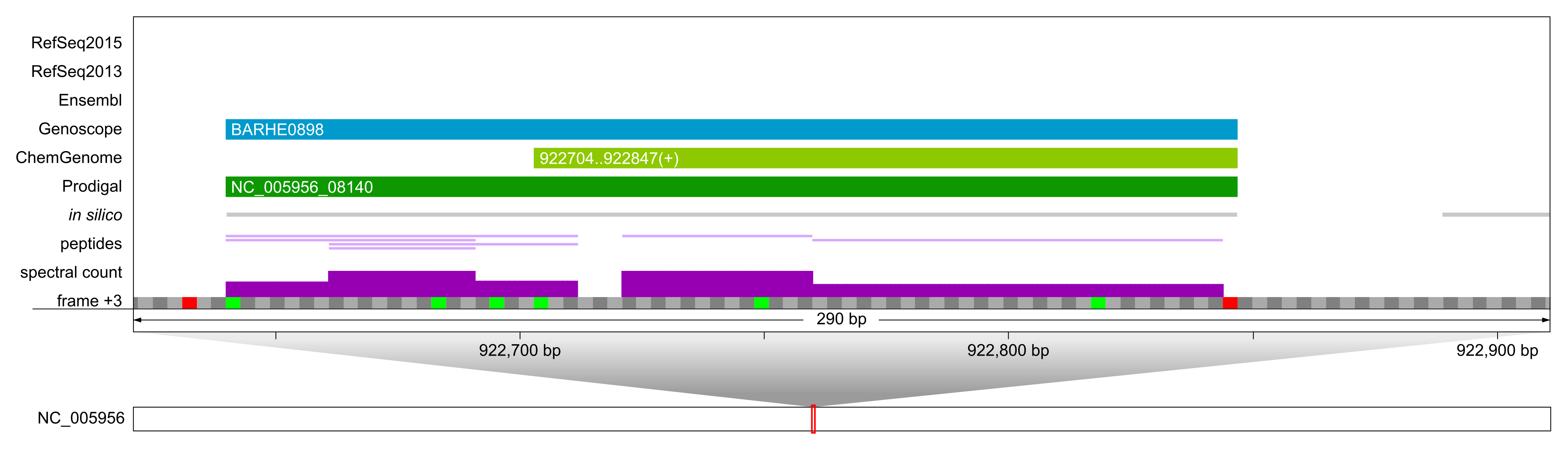
Example of a novel, differentially expressed sORF of 68 amino acids (BARHE0898, frame +3), that was expressed roughly 6.5-fold higher in the induced condition [1]. Peptide evidence supported the longer form of this ORF annotated by Genoscope and Prodigal.
Example of a novel, in silico only predicted sORF of 34 aa. This particular sORF was identified with one single peptide and 4 PSMs and does not overlap with any other annotation. Its expression could be confirmed by PRM; our predominant SCL computation [2] indicated that it is localized to the inner membrane.

A highly expressed pseudogene (RefSeq2015: BH_RS01070, frame -3) annotated as normal CDS (transcription elongation factor NusA) by RefSeq2013, Ensembl, and Genoscope was identified (2244 spectra are mapped to 117 peptides of NusA), which is annotated as pseudogene in RefSeq2015 for unknown reason. There is no experimental evidence for the +8 aa N-terminal extension predicted by ChemGenome.

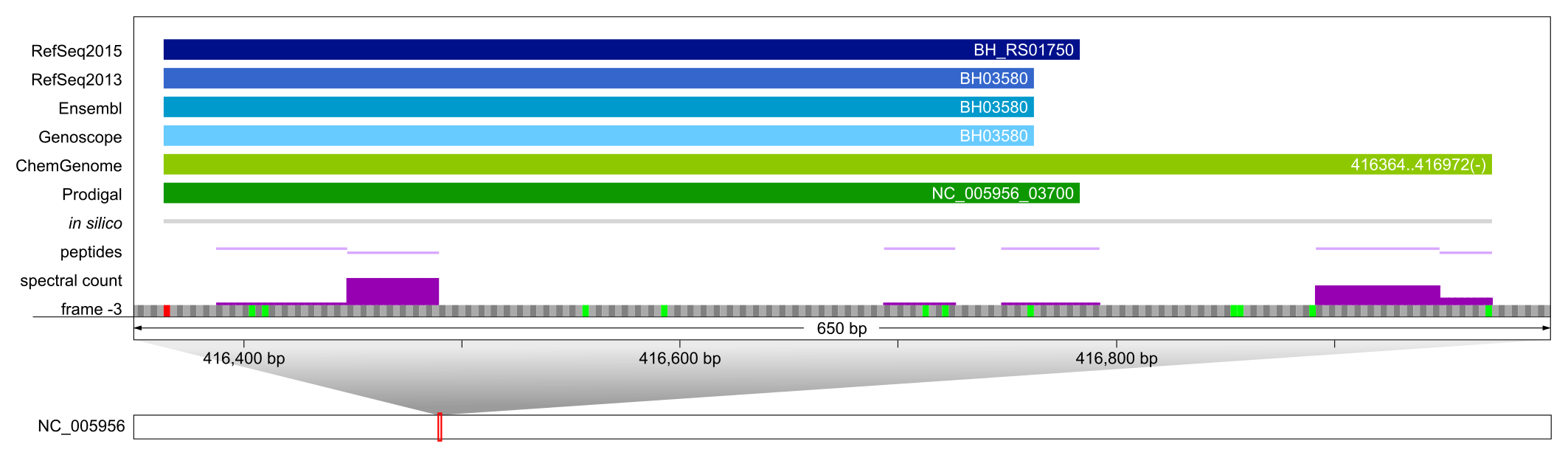
For BH_RS01750, a hypothetical protein encoded in a prophage region, only ChemGenome correctly predicted a 63 aa longer proteoform than annotated by other resources; its expression was supported by several peptides.
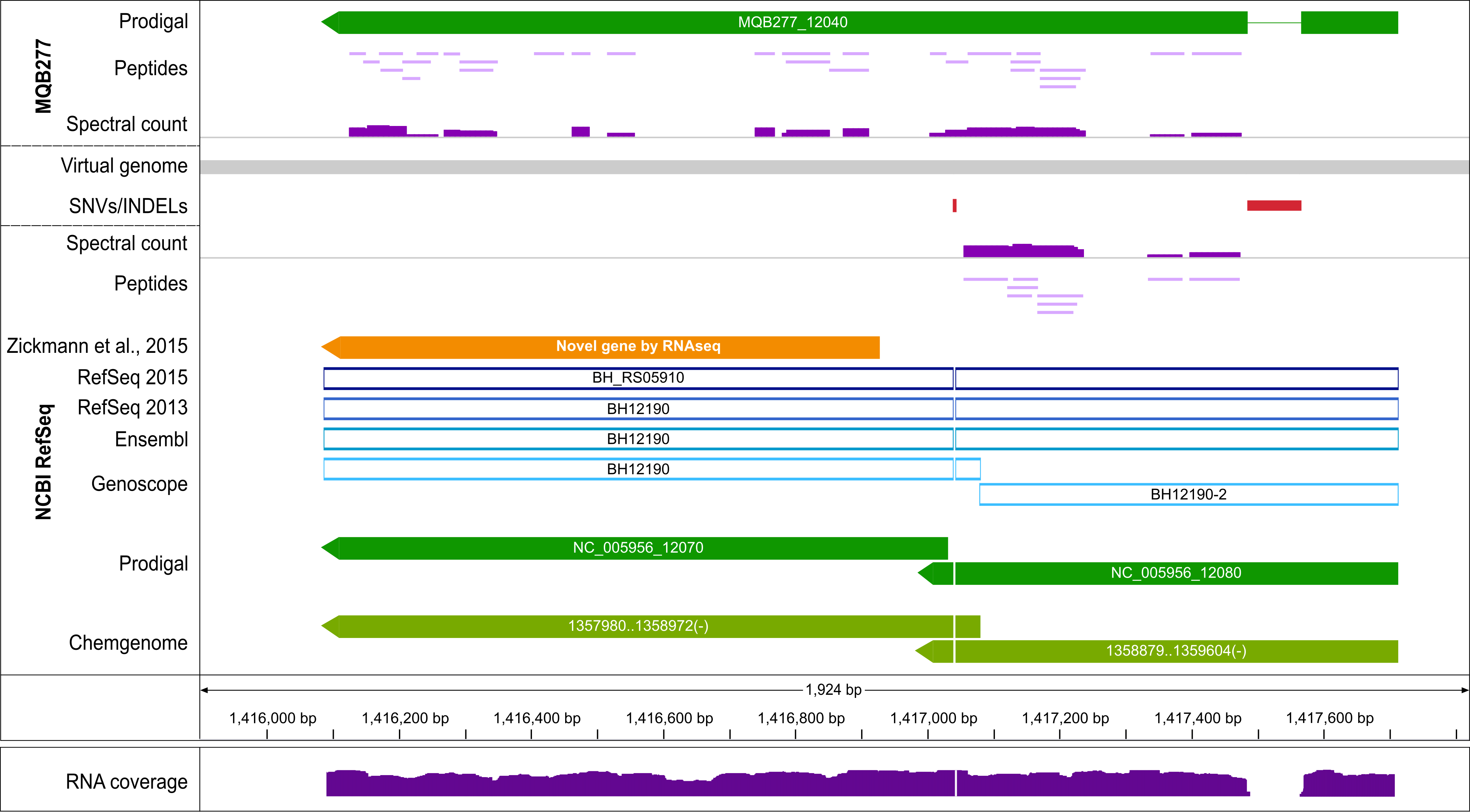
Genomic region encoding an ABC transporter (BH_RS05910). RefSeq and Ensembl
annotate it as pseudogene, Genoscope as fragmented pseudogene, while Prodigal and Chemgenome predict
2 CDSs. The reference genome (below the gray virtual genome bar; NCBI
RefSeq track) differs from the assembly of the actual Bartonella henselae laboratory strain
MQB277 [3] (MQB277 track above the virtual genome) by an insertion of 81 bp and a
1 bp deletion (red
boxes); the 1 bp deletion causes a frameshift, evidenced by the lack of protein expression
downstream of it (spectral count below the virtual genome; scaled from 0 to 800) and by
transcriptomic data (reads mapped to the reference genome all support the insertion; lower panel).
In contrast, the protein encoded by MQB277_12040 in the assembly is expressed over almost its entire
length (class 1a peptides; 1 peptide identified by 7 PSMs spans the
frameshift region), also supported by transcriptomic reads mapping without any mismatch (data not
shown).
An RNA-seq based proteogenomics study [4] identified a novel gene when mapping our
transcriptomics and proteomics data [1] against the NCBI reference genome
(orange bar).
However, the ORF is only accurately identified accurately by having the
correct genome sequence available.
Evidence for a SNV causing a non-synonymous SAAV in the CDS of transcription elongation factor GreA. Four peptides (2, 4, 8, 39 PSMs) confirming this SAAV (Glycine in reference to Glutamic acid in our assembly) are mapped to this position in MQB277.
