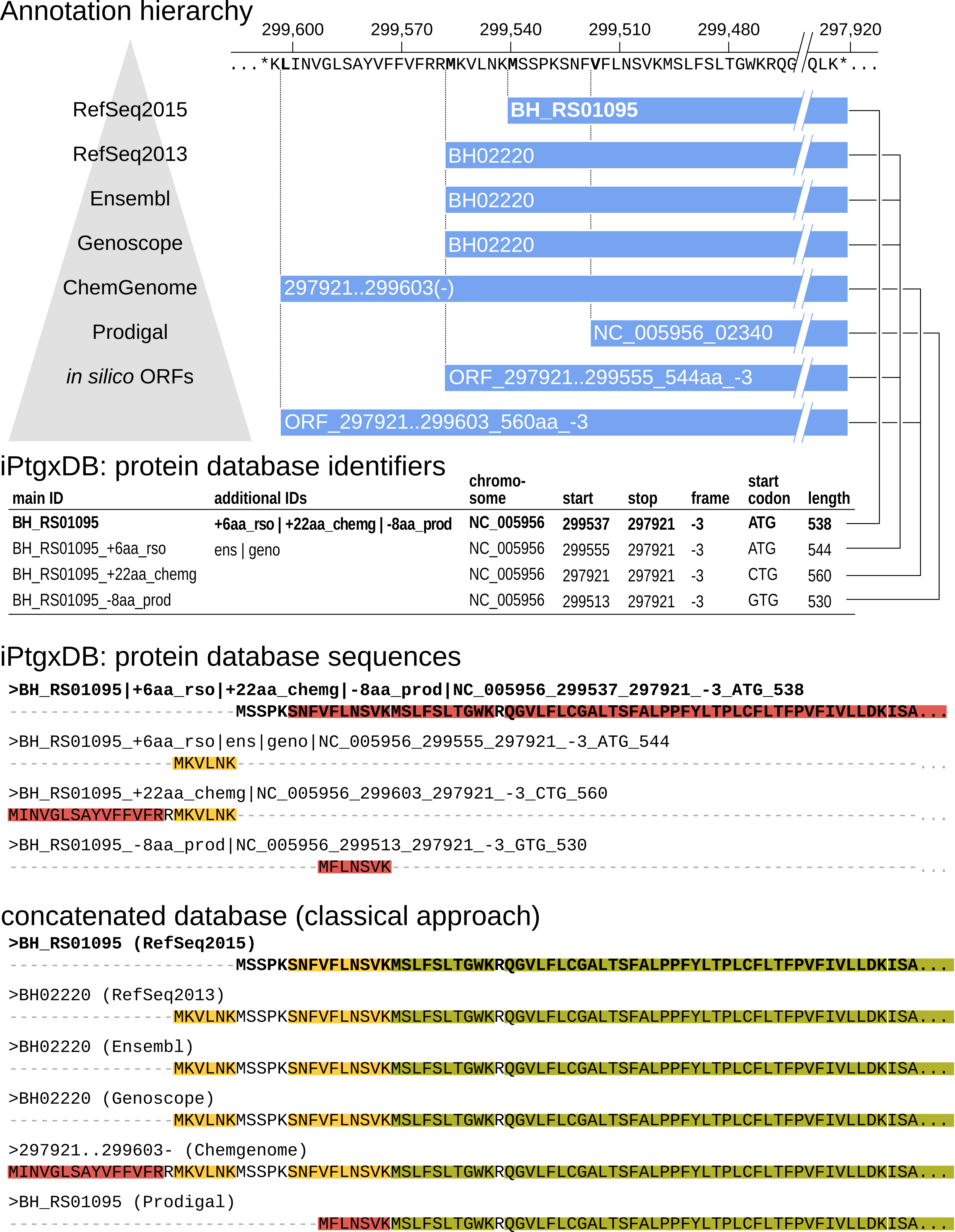
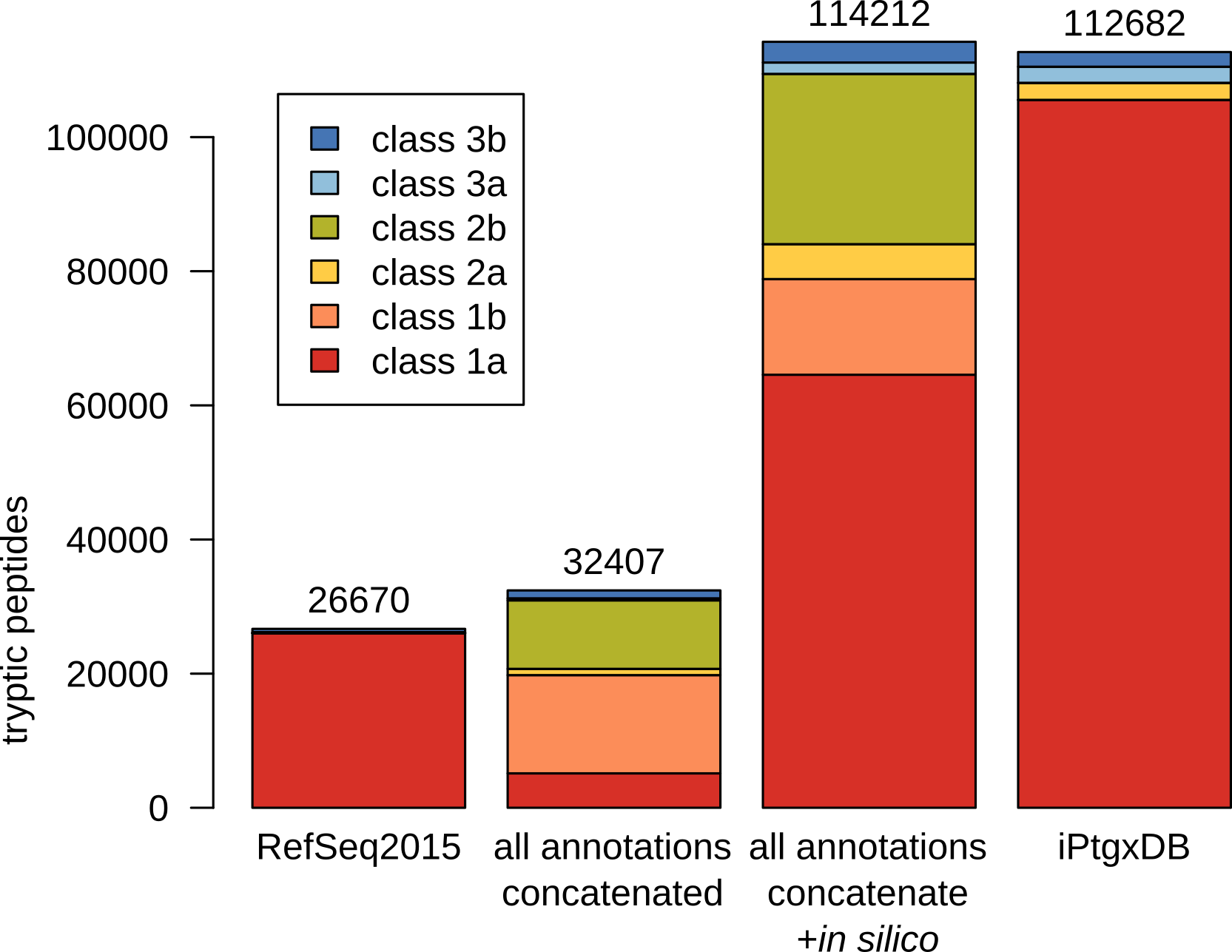
In this example, CDSs and pseudogenes of 7 resources are integrated in a stepwise fashion, following a hierarchy: to leverage the quality of manual curation efforts we start with reference genome annotations, then ab initio predictions, then in silico ORFs from a six-frame translation. A unique aspect of our iPtgxDBs is that almost all peptides uniquely identify one specific protein. This was accomplished by extending the PeptideClassifier concept [1] for prokaryotes [2]. Our extension treats protein sequences with a common stop codon and varying start positions (N-termini) as a protein annotation cluster, i.e. variants of a prokaryotic gene model (similar to isoforms of a eukaryotic gene model). The anchor sequence for an annotation cluster is selected from the annotation highest up in the hierarchy, i.e. here RefSeq2015, unless no CDS is predicted in a given genomic region.
Informative protein identifiers are created (Interpreting identifiers), illustrated for the annotation cluster with the RefSeq2015 anchor sequence BH_RS01095 shown in bold, where three additional start sites exist. The four different proteoforms are added to the protein search DB: the anchor sequence (bold) with the full protein sequence, the extensions (RefSeq2013 and ChemGenome) add the upstream sequence up to the first tryptic cleavage site within the anchor sequence. The shorter Prodigal prediction uses an alternative start codon resulting in a distinguishable N-terminal peptide, and therefore gets also added. The two in silico ORFs are identical to annotations higher up in the annotation hierarchy. Peptide classes are shown for the N-terminal sequences of the CDS annotation cluster (see also Fig. 3B).