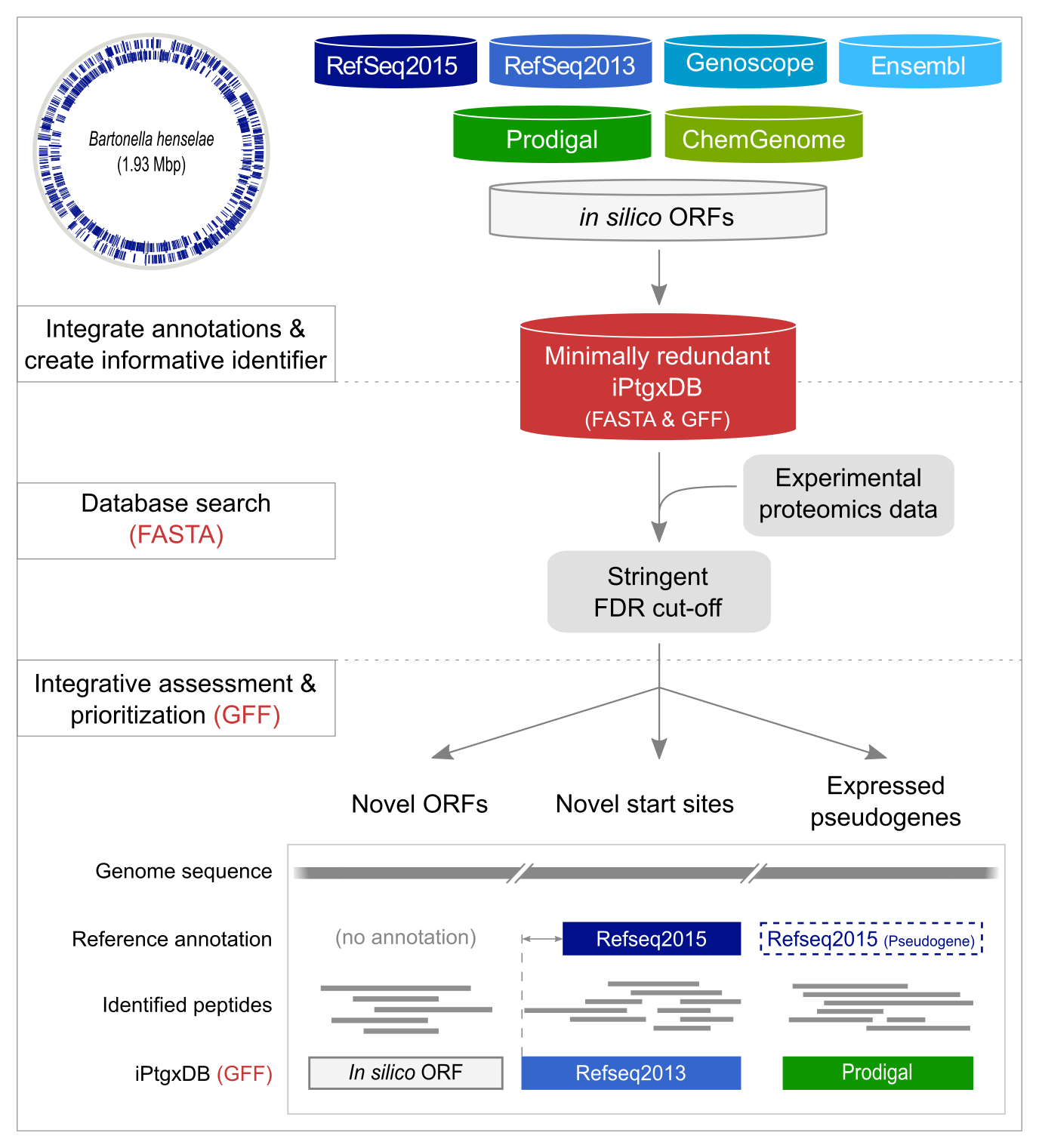
For a completely sequenced prokaryotic genome (B. henselae strain Houston-1 is shown as example with annotated CDSs), reference genome annotations (blue containers), results from ab initio gene prediction algorithms (green containers), and in silico ORFs (white container), are downloaded or computed and integrated in a first pre-processing step (upper panel). All CDS and pseudogene annotations are matched, informative gene identifiers are created and stored in a minimally redundant iPtgxDB (red container). iPtgxDBs come in the form of searchable protein sequences in FASTA format and integrated annotations as a GFF file.
Experimental proteomics data are matched to the DB using a target-decoy approach relying on stringent FDR cut-offs (middle panel). We have made very good experiences with the MS-GF+ software [1].
Identified PSMs and peptides are post-processed to visualize novel candidates like novel ORFs, novel start sites or expressed pseudogenes (lower panel) (Examples of uncovered novelties) in the context of experimental data integrated with the GFF file.